Tutorial: using NeurEco python API on a Tabular Classification problem
Contents
Tutorial: using NeurEco python API on a Tabular Classification problem#
The following section uses the test case Gene expression cancer RNA sequence. This test case is included in the NeurEco installation package.
Create an empty directory (geneCancer Example), extract the Gene expression cancer RNA sequence test case data there. The created directory contains the following files:
x_test.csv
y_test.csv
x_train_0.csv
y_train_0.csv
x_train_1.csv
y_train_1.csv
Build a model#
Import the required libraries (NeurEco and NumPy):
from NeurEco import NeurEcoTabular as Tabular
import numpy as np
Load the training data:
x_train = []
y_train = []
for i in range(2):
x_name = "x_train_" + str(i) + "_.csv"
y_name = "y_train_" + str(i) + "_.csv"
x_part = np.genfromtxt(x_name, delimiter=";", skip_header=True)
x_train.append(x_part)
y_part = np.genfromtxt(y_name, delimiter=";", skip_header=True)
y_train.append(y_part)
x_train = np.vstack(tuple(x_train))
y_train = np.vstack(tuple(y_train))
Initialize a NeurEco object to handle the Classification problem:
builder = Tabular.Classifier()
All the methods provided by the Classifier class, can be viewed by calling the __method__ attributes:
print(builder.__methods__)
*** NeurEco Tabular Classifier methods: ***
- load
- save
- delete
- evaluate
- build
- get_input_count
- get_output_count
- load_model_from_checkpoint
- get_number_of_networks_from_checkpoint
- get_weights
- export_fmu
- export_c
- export_onnx
- export_vba
- compute_error
- plot_network
- forward_derivative
- gradient
- set_weights
- perform_input_sweep
To understand what each parameter of any method does and how to use it print the doc of the method:
print(builder.export_c.__doc__)
exports a NeurEco tabular model to a header file
:param h_file_path: path where the .h file will be saved
:param precision: string: optional: "float" or "double": precision of the weights in the h file
:return: export_status: int: 0 if export is ok, other if otherwise.
To build the model, run the build method with the building parameters adjusted to the problem at hand (see Build NeurEco Classification model with the Python API). For this example, the outputs to be normalized per feature (meaning that each output will be normalized apart, it is the default setting for Compression, see Data normalization for Tabular Compression):
builder.build(input_data=x_train, output_data=y_train,
# the rest of these parameters are optional
write_model_to="./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.ednn",
checkpoint_address="./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.checkpoint",
valid_percentage=33.33)
When build is called, NeurEco starts the building process:
Validation Percentage will be used to get the validation data. This is due to:
- one or all the validation data is set to None
- validation indices is set to None
info >
info > _ __ ______
info > / | / /__ __ _______/ ____/________
info > / |/ / _ \/ / / / ___/ __/ / ___/ __ \
info > / /| / __/ /_/ / / / /___/ /__/ /_/ /
info > /_/ |_/\___/\__,_/_/ /_____/\___/\____/
info > === A D A G O S ===
info >
info > Version: 4.01.2474.0 Compiled with MSVC v1928 Oct 12 2022 Matlab runtime:no
info > OpenMP: yes
info > MKL: yes
info > Reading data files...
info > Reading Data from C:/Users/Sadok/AppData/Local/Temp/tmplluno8ip/inputs_tab_train.npy
info > Reading Data from C:/Users/Sadok/AppData/Local/Temp/tmplluno8ip/outputs_tab_train.npy
info > build for: 5 outputs and 20531 inputs and 640 samples.
info > Preparing Inputs
info > Building Model
During the build NeurEco saves the intermediate modes to the checkpoint file (defined by the parameter checkpoint_address). To load and use the intermediate models from this checkpoint:
Create a new NeurEco object in which to load the model:
model = Tabular.Classifier()
Determine how many intermediate models the checkpoint contains:
n = model.get_number_of_networks_from_checkpoint("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.checkpoint")
Load any intermediate model from the checkpoint using its id (count starts with zero). For this example, at the moment of running the command \(n=6\) and the following command loads the intermediate model \(n°3 \ (id=2)\):
model.load_model_from_checkpoint("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.checkpoint", 2)
Now model is a valid Compression model, and can be used as usual.
Check the number of trainable parameters each of the intermediate models has:
for i in range(n):
print("Loading model", i, " from checkpoint file:")
model.load_model_from_checkpoint("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.checkpoint", i)
print("number of trainable parameters in intermediate model --", i, " is:", model.get_weights().size)
Loading model 0 from checkpoint file:
number of trainable parameters in intermediate model -- 0 is: 157
Loading model 1 from checkpoint file:
number of trainable parameters in intermediate model -- 1 is: 157
Loading model 2 from checkpoint file:
number of trainable parameters in intermediate model -- 2 is: 148
Loading model 3 from checkpoint file:
number of trainable parameters in intermediate model -- 3 is: 148
Loading model 4 from checkpoint file:
number of trainable parameters in intermediate model -- 4 is: 148
Loading model 5 from checkpoint file:
number of trainable parameters in intermediate model -- 5 is: 148
Evaluate a model#
Load the testing data from the CSV files:
x_test = np.genfromtxt("x_test.csv", delimiter=";", skip_header=True)
y_test = np.genfromtxt("y_test.csv", delimiter=";", skip_header=True)
Create a Classifier object to use for the evaluation:
evaluator = Tabular.Classifier()
Note
It is possible to use the already existing Classifier object builder when the evaluation is done just after the build, and builder is still available.
Load the built model:
load_state = evaluator.load("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq")
Note
When building or evaluating a NeurEco model, all the used paths don’t necessarily need to have an extension when it is passed as a parameter to a NeurEco method, being for a model or for a checkpoint file.
To extract information from the loaded model, such as the number of inputs, the number of outputs and the weights array, run:
n_inputs = evaluator.get_input_count()
n_outputs = evaluator.get_output_count()
weights = evaluator.get_weights()
print("Number of Inputs:", n_inputs)
print("Number of Outputs:", n_outputs)
print("Number of trainable parameters:", weights.size)
Number of Inputs: 20531
Number of Outputs: 5
Number of trainable parameters: 148
To plot the network graph (this operation requires matplotlib library installed, see Plot a NeurEco network):
evaluator.plot_network()
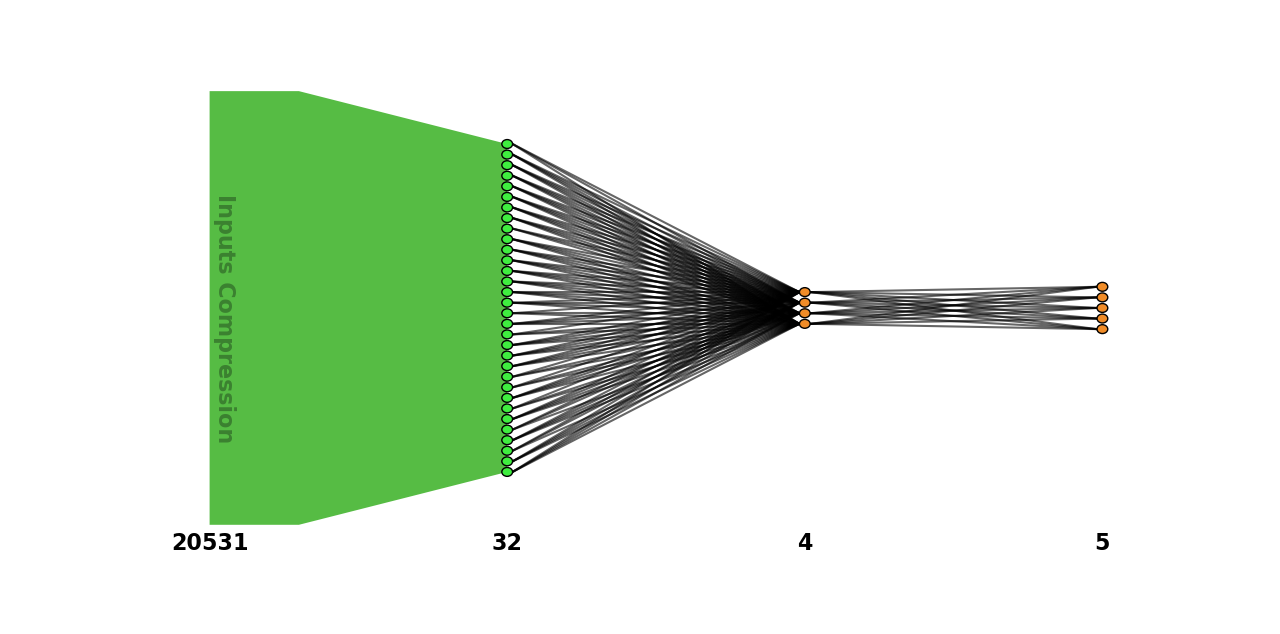
Python API operations: plotting a network: test case - GeneCancer#
To evaluate the model on the test data:
neureco_outputs = evaluator.evaluate(x_test)
l2_error = evaluator.compute_error(neureco_outputs, y_test)
print("L2 relative error (%):", 100 * l2_error)
L2 relative error (%): 0.0
Note
During evaluation, the normalization is carried out by the model and its parameters are not relative to the data set being evaluated, but are the global parameters computed during the build of the model.
To save the model in the native NeurEco binary format:
save_state = evaluator.save("GeneExpressionCancerRnaSeqModel//NewDir/SameModel")
To export the model, run one of the following commands (embed license is required):
save_state = evaluator.save("GeneExpressionCancerRnaSeqModel//NewDir/SameModel")
evaluator.export_c("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.h", precision="float")
evaluator.export_onnx("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.onnx", precision="float")
evaluator.export_fmu("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.fmu")
evaluator.export_vba("./GeneExpressionCancerRnaSeqModel/GeneExpressionCancerRnaSeq.bas")
Warning
Once the NeurEco object is no longer needed, free the memory by deleting the object by calling the delete method. For the example above, three objects must be deleted:
builder.delete()
evaluator.delete()
model.delete()